What is Common Technical Document Module 2.5?
The Common Technical Document (CTD) is a standardised format for preparing application dossiers for the registration of investigational medicinal products (IMPs) in participating regions including the EU, Japan, and the US [1]. The CTD is a large, multidisciplinary assembly of documents comprising five modules, into which all quality, safety, and efficacy information on the IMP is compiled.
Here, we will focus on Module 2.5, the Clinical Overview (CO) [1].
Figure 1. Common Technical Document triangle
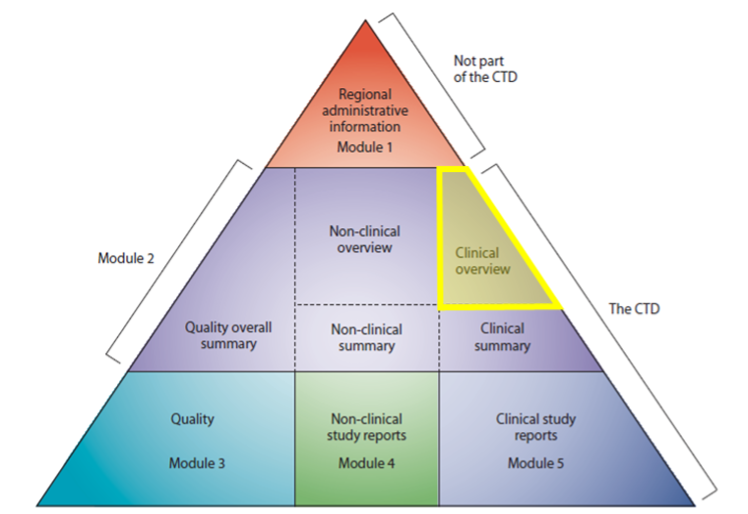
CTD = Common Technical Document.
The CTD is organised into five modules. Module 1 is region-specific and Modules 2‑5 are intended to be common for all regions [1].
Module 2.5, the CO, provides reviewers with a critical analysis of the clinical data available for an IMP. Specifically, it contains a succinct discussion and interpretation of clinical information along with any other relevant information with important clinical implications and thus supports the proposed product label [2].
What are the main requirements for a CO and how can Alchemy help?
At Alchemy, we have extensive experience in authoring clinical CTD modules. The key requirements for developing a comprehensive and effective CO include:
1. Adherence to regulatory guidelinesummary (PLS)
We ensure your CO adheres to regulatory guidelines outlined by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and apply industry best practices to ensure consistency and compliance. The relevant general ICH guidelines for the CO include the following:
- ICH M4 (R4): Organisation of the CTD for the registration of pharmaceuticals for human use
- ICH M4 (R3): Questions and answers: Organisation of the CTD for the registration of pharmaceuticals for human use
- ICH M4E (R2): Guideline on enhancing the format and structure of benefit-risk information in ICH – efficacy
- ICH M4E (R4): Questions and answers: Guideline on enhancing the format and structure of benefit-risk information in ICH – efficacy
2. Module 2.5 content coordination
A standalone peer-reviewed article summarising a previously published article, using non-technical language and graphic design to enhance visual communication.
3. Document publishing
We can perform submission-ready publishing of documents ahead of electronic CTD collation and publishing, which would be performed by a submission publisher.
4. Streamlining of entire CTD document
We offer streamlined writing support across all CTD modules to apply consistency, style preferences, and regulatory adherence across the CTD.
How Module 2.5 is used and its role within the regulatory framework
The primary role of the CTD is to provide a standardised format for preparing marketing authorisation applications, new drug applications, and biologic license applications in participating regions [4-6]. The CTD is mandated or strongly recommended in several regions including Europe, the US, Japan, Canada, China, and Switzerland [1,7,8].
Module 2.5, the CO, provides an overview and critical analysis of the clinical information available for an IMP. It is ideally a short document of approximately 30 pages which, alongside the Clinical Summary (Module 2.7), provides the supporting information for the Summary of Product Characteristics (SmPC), United States prescribing information (USPI), or other product label. The CO is structured into six sections, as follows, to guide the reviewer through interpretation of the clinical data:
- Product development rationale
- Biopharmaceutics
- Clinical pharmacology
- Efficacy
- Safety
- Benefit/risk conclusions
To aid the reviewer’s understanding of the clinical data, the CO should contextualise findings by referencing relevant literature and explaining how the IMP would fit into the existing clinical landscape following approval [2]. Preparation of a clear, well-informed CO that maintains consistency with the rest of the CTD is therefore essential to support applications for marketing authorisation of an IMP in participating regions [7].
Which source materials are needed to write Module 2.5?
To develop a comprehensive and high-quality CO, you would need to provide a range of source documents including, but not limited to:
- Clinical Study Reports (CSRs)
- Module 2.7 Clinical Summary documents (or drafts there-of)
- Proposed product label
- Proposed risk management plan (RMP) or risk evaluation and mitigation strategies (REMS)
- Regulatory briefing documents and responses or advice from regulatory agencies (as appropriate for the submission)
- Investigator’s Brochure (IB)
- Investigational Medicinal Product Dossier, if no is IB available
- Any relevant literature
- Sponsor style guide
- Other materials for appendices
What is the general timeframe for writing Module 2.5?
The timeframe for preparation of the CO is typically a few months [9,10]. Timelines vary depending on the complexity of the submission, for example the number of indications being investigated and the volume of clinical data available [2]. Effective team alignment and communication from project kick-off will accelerate CO development.
Overview of the development of CTD Module 2.5, the Clinical Overview [10]

CO = Clinical Overview; CTD = Common Technical Document; QC = Quality control; SME = subject matter expert.
Alchemy’s top tips for authoring CTD Module 2.5?
Crafting a high-quality CO is critical for regulatory success. It’s your opportunity to tell the scientific story of your IMPs risk/benefit profile in a concise, persuasive, and evidence-based way. Below are some top tips to help you develop an impactful CO.
1. Start with the purpose in mind
The CO is an interpretation of clinical data. It should:
- Integrate and critically assess all clinical information (Modules 2.7 and 5)
- Explain the clinical development strategy and rationale
- Articulate the risk/benefit assessment clearly and convincingly
- Provide a narrative bridge between data and decision-making for regulators
If you were a regulator reading this for the first time, would you be convinced that the IMPs benefits outweigh its risks?
2. Follow the structure, but tell a story
Whilst ICH guidelines define the required sections, the best documents provide a logical, readable story. Use transition sentences to connect sections and maintain narrative flow.
3. Be interpretive, not repetitive
Don’t just restate results from the Clinical Summary or CSRs. Instead:Interpret trends, relationships, and consistencies across studies
Highlight key differentiators versus existing therapies
- Explain anomalies or unexpected findings transparently
- A few clear visuals such as forest plots and risk/benefit matrices can make a big impact
4. Address safety proactively
Regulators look for transparency and critical thinking. Present safety data systematically and identify knowledge gaps, showing how they’re being mitigated (e.g., post-marketing surveillance or ongoing studies, or through prescribing restrictions as described in the RMP or REMS). It is best to acknowledge limitations and show proactive management.
5. Present the risk/benefit clearly
This is the heart of the CO. Use a structured approach, balancing patient-centric outcomes with clinical data. Provide clear justification for why benefits outweigh risks for the intended population. Conclude with confidence but support conclusions with logic and data.
6. Ensure consistency across CTD modules
Regulators will cross-check statements between Module 2.5 (CO), Module 2.7 (Clinical Summary), and Module 5 (CSRs). It is therefore important to keep data consistent (no discrepancies in numbers or conclusions), cross-reference rather than repeat details, and ensure terminology, units, and definitions are harmonised. Developing a company style guide early in the clinical development process can reduce time spent harmonising documents later.
7. Write for readability and impactAim for clarity and precision using short, active sentences, and placing key messages early in paragraphs and sections. Keep the tone professional.
8. Include a clear and balanced conclusion
End with a succinct, evidence-based statement that reinforces:
- The IMPs overall clinical value
- The target population most likely to benefit
- Any risk management strategies to ensure safe use
The CO is your scientific argument. It should be a coherent, evidence-driven narrative, demonstrating full understanding of your data and the disease under study. It should provide insight into clinical relevance of your IMP in the target population.
9. Peer review and quality control
Have independent reviewers (medical, statistical, regulatory) critique the document. Perform a complete quality control check for data accuracy and compliance.
10. Final thoughts
Here’s what our Managing Director, Dr Adam Taylor, had to say about COs:
“A high-quality Clinical Overview isn’t just a summary of studies; it’s the product’s scientific narrative. It must interpret data, connect the evidence, and clearly demonstrate why the benefits of the product outweigh its risks. The best overviews are transparent, data‑driven, and tell a compelling, credible story that inspires regulatory confidence.”
How can I contact Alchemy about writing CTD Module 2.5?
To discuss your CTD writing needs with our team, please contact us here.
You can read more about our experience with CTDs here and here
Sources:
- ICH Official web site: ICH
- ICH M4E Common technical document for the registration of pharmaceuticals for human use – efficacy – Scientific guideline | European Medicines Agency (EMA)
- ICH: multidisciplinary | European Medicines Agency (EMA)
- eCTD guidance for marketing authorisation and post-authorisation applications – GOV.UK
- Submitting Marketing Applications According to the ICH/CTD Format: General Considerations | FDA
- The Biologics License Application (BLA) in Common Technical Document (CTD) Format – Vaccine Development and Manufacturing – Wiley Online Library
- https://journal.emwa.org/regulatory-writing-basics/an-overview-of-the-common-technical-document-ctd-regulatory-dossier/article/1693/2047480614z2e000000000207.pdf
- CDE Consults on the Structure of Electronic Common Technical Document (eCTD) for Drug Registration
- Step-by-Step Guide to Drug Dossier Preparation for Approval
- Preparation of CTD modules for an IND application – Alchemy Medical Writing